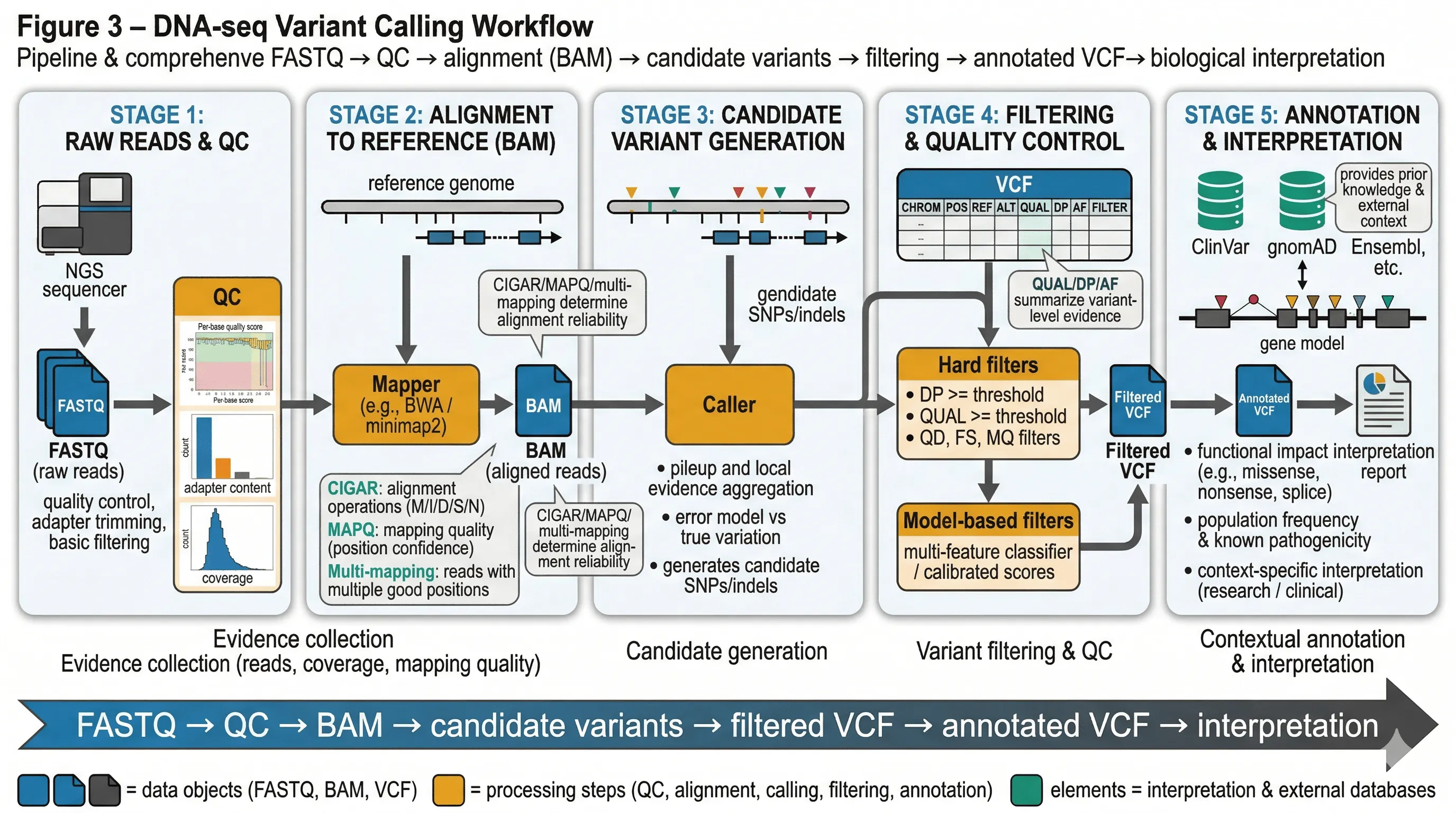
DNA-seq 变异检测总览
这是一页把 DNA-seq variant calling 放回完整分析链路的概览:你需要同时看 BAM、VCF、参考版本、过滤逻辑和证据质量,而不是把 caller 当成黑箱。
- 如果你刚开始接触变异检测,先把 FASTQ → BAM → VCF 的层级关系理顺。
- 这页重点是流程和判断逻辑,不是具体软件命令。
DNA-seq 变异检测要解决的核心问题是:
输入:参考基因组序列 (如 GRCh38),以及来自待测样本的比对后测序数据(BAM/CRAM 格式)。 输出:一组候选变异位点,每个位点包含参考等位基因(REF)、替代等位基因(ALT)、质量分数(QUAL)、以及支持证据的统计摘要。
该问题的本质是在观测到的序列差异中,区分真实的生物学变异与技术噪声、比对假象、测序错误。
变异检测不是单纯”找不同”,而是要在技术噪声和真实信号之间做区分。一个可靠的变异检测流程需要同时考虑:
- 数据层级:FASTQ 中的原始碱基质量、BAM 中的比对证据、VCF 中的变异质量分数之间的层级关系;
- 比对不确定性:重复区域的多重比对、indel 周围的局部重比对失败如何影响变异判断;
- 统计建模:测序深度的泊松波动、碱基错误率的系统偏差、strand bias 等偏好性;
- 过滤逻辑:硬阈值过滤与机器学习模型过滤的权衡;
- 注释与解释:数据库版本、参考基因组版本、功能注释对下游分析的影响。
如果忽略这些层次,就很容易把 caller 的输出误当成”自动得到的真变异”,导致后续分析建立在不可靠的候选集上。
1. 原始数据与质量控制
Section titled “1. 原始数据与质量控制”变异检测的可靠性首先取决于输入数据的质量。从 FASTQ 出发,需要系统性地评估:
- 碱基质量分数(Phred score):,反映每个碱基的测序可信度;
- 接头与引物污染:文库构建过程中引入的外源序列需要在比对前去除;
- 覆盖度分布:理想情况下,基因组各区域的测序深度应服从泊松分布,异常的高覆盖或低覆盖区域可能提示技术偏好或样本问题;
- 批次效应与样本污染:不同测序批次或混合样本会引入系统性偏差。
低质量输入会直接影响后续候选位点的证据强度,导致假阳性或假阴性。
2. 比对到参考基因组
Section titled “2. 比对到参考基因组”变异检测高度依赖比对(alignment)的质量。比对问题主要包括:
- 重复区域的多重比对(multi-mapping):短 reads 在基因组重复区域可能匹配到多个位置,导致 MAPQ 降低,变异判断困难;
- Indel 周围的局部比对失败:标准 Smith-Waterman 局部比对在 indel 边界处容易产生错位,需要局部重比对(local realignment);
- 参考基因组版本不一致:使用不同参考版本(如 GRCh37 vs GRCh38)会导致坐标偏移,使”变异”实际上只是坐标系差异。
3. 候选位点生成
Section titled “3. 候选位点生成”变异检测算法(caller)的核心任务是在以下因素之间做统计区分:
| 信号来源 | 特征 | 判别方法 |
|---|---|---|
| 真实变异 | 等位基因频率符合孟德尔定律或肿瘤克隆结构;支持 reads 质量高 | 贝叶斯概率模型、基因型似然比 |
| 测序错误 | 随机分布;低碱基质量;通常仅出现在单条 read | 错误率建模、质量分数校准 |
| PCR 重复 | 相同起始位置的 reads 过多;偏好性扩增 | 去重(deduplication)、分子标签(UMI) |
| 比对假象 | 集中在重复区域;MAPQ 低;CIGAR 复杂 | 多重比对过滤、重复区域屏蔽 |
4. 过滤与注释
Section titled “4. 过滤与注释”最终 VCF 中的 FILTER 字段和 QUAL 字段是 caller 对候选位点可信度的量化。分析者需要理解:
- FILTER 标签的含义:PASS 表示通过所有内部过滤;其他标签(如 LowQual、StrandBias)提示特定问题;
- QUAL 分数的统计意义:通常是 Phred 标度的变异为假阳性的概率;
- INFO 字段的解读:DP(深度)、AF(等位基因频率)、SB(链偏好)等统计量帮助人工审核;
- 注释一致性:功能注释(如 VEP、SnpEff)依赖于转录本数据库版本,需与参考基因组版本匹配。
一个简化 DNA-seq variant workflow 可以写成:
FASTQ -> QC -> alignment -> BAM -> candidate variants -> filtering -> VCF annotation -> interpretation如果某个候选 SNP 只被极少数 reads 支持,而且这些 reads 的位置质量较差,那么它更可能是:
- 测序错误;
- 比对错位;
- 低复杂度区域产生的假阳性。
因此变异检测的关键并不是”有差异就算变异”,而是评估证据质量。
与真实工具或流程的连接
Section titled “与真实工具或流程的连接”- 比对:索引结构、seed-and-extend、动态规划;
- 候选位点生成:局部证据聚合、错误建模;
- 过滤:统计阈值、经验规则、群体频率与功能注释;
- 解释:数据库映射、转录本 / 蛋白水平影响分析。