RNA-seq 工作流概览
这是一页把 RNA-seq 从原始 reads 一路串到定量、归一化、差异分析与功能解释的总览页面,重点是建立流程地图而不是记命令。
- 如果你刚开始接触 RNA-seq,先把流程中的输入输出、注释依赖和定量目标区分清楚。
- 如果你已经做过一些分析,这页更适合你回头定位每一步依赖的算法与统计前提。
RNA-seq 工作流描述的是:如何从转录组测序得到的 reads 出发,经过比对或 pseudo-alignment、表达定量、统计分析与功能解释,最终回答基因表达和转录调控相关问题。
它既是 NGS 工作流中的一个重要分支,也是很多生物学研究里最常见的分析路线之一。
RNA-seq 常用于回答:
- 哪些基因在样本中被表达;
- 表达量有多高;
- 不同条件之间是否存在差异表达;
- 是否存在新的剪接事件或转录本结构。
相比 DNA 测序,RNA-seq 的一个关键特点是:
- 它面对的是转录本而不是整个基因组;
- 同一个基因可能有多个 isoform;
- 结果很依赖注释和定量模型。
1. 质控与过滤
Section titled “1. 质控与过滤”与一般 NGS 一样,RNA-seq 也要先检查:
- read 质量是否稳定;
- 是否有接头污染;
- 是否存在样本间明显偏差;
- 文库复杂度是否足够。
但 RNA-seq 还需要特别警惕:
- rRNA 污染;
- 3’ 偏好或覆盖不均;
- 批次效应。
2. 比对或 pseudo-alignment
Section titled “2. 比对或 pseudo-alignment”RNA-seq 的核心选择之一是:
- 做传统 spliced alignment;
- 或做 pseudo-alignment / lightweight mapping。
前者更适合需要精确定位、剪接分析或新转录本研究的场景;后者在表达定量任务中通常更快。
这里隐含的算法问题包括:
- 如何处理跨外显子 reads;
- 如何在多个转录本之间分配 read;
- 如何使用索引结构快速定位候选位置。
定量的目标是估计:
- 基因层表达量;
- 转录本层表达量。
但定量并不是简单计数。困难在于:
- 一个 read 可能同时兼容多个 isoform;
- 长基因和短基因的计数不可直接比较;
- 总测序深度不同也会影响原始 counts。
因此,定量通常结合模型、有效长度和多重比对分配策略来完成。
4. 归一化
Section titled “4. 归一化”归一化是为了让不同样本之间的表达值可以更公平地比较。
常见动机包括:
- 测序深度不同;
- 文库组成差异;
- 某些高表达基因对整体分布产生偏移。
5. 差异分析
Section titled “5. 差异分析”差异分析要回答的是:
- 某个基因在条件 A 和条件 B 下是否显著不同;
- 变化方向和幅度是什么;
- 统计显著性是否足够。
这里常涉及:
- count model;
- dispersion estimation;
- multiple testing correction。
6. 功能解释
Section titled “6. 功能解释”仅有差异表达基因列表通常还不够,我们还需要进一步做:
- GO / pathway enrichment;
- gene set interpretation;
- 与数据库和文献结果对照;
- 与实验背景结合解释。
一个简化的 RNA-seq 分析链路可以写成:
FASTQ -> QC -> spliced alignment / pseudo-alignment -> quantification -> normalization -> DE analysis -> enrichment如果最后差异表达结果看起来很奇怪,问题可能不只在统计模型,也可能出在:
- 样本分组设计;
- 质控不足;
- 注释版本不匹配;
- 多重比对 read 的处理方式。
与真实工具或流程的连接
Section titled “与真实工具或流程的连接”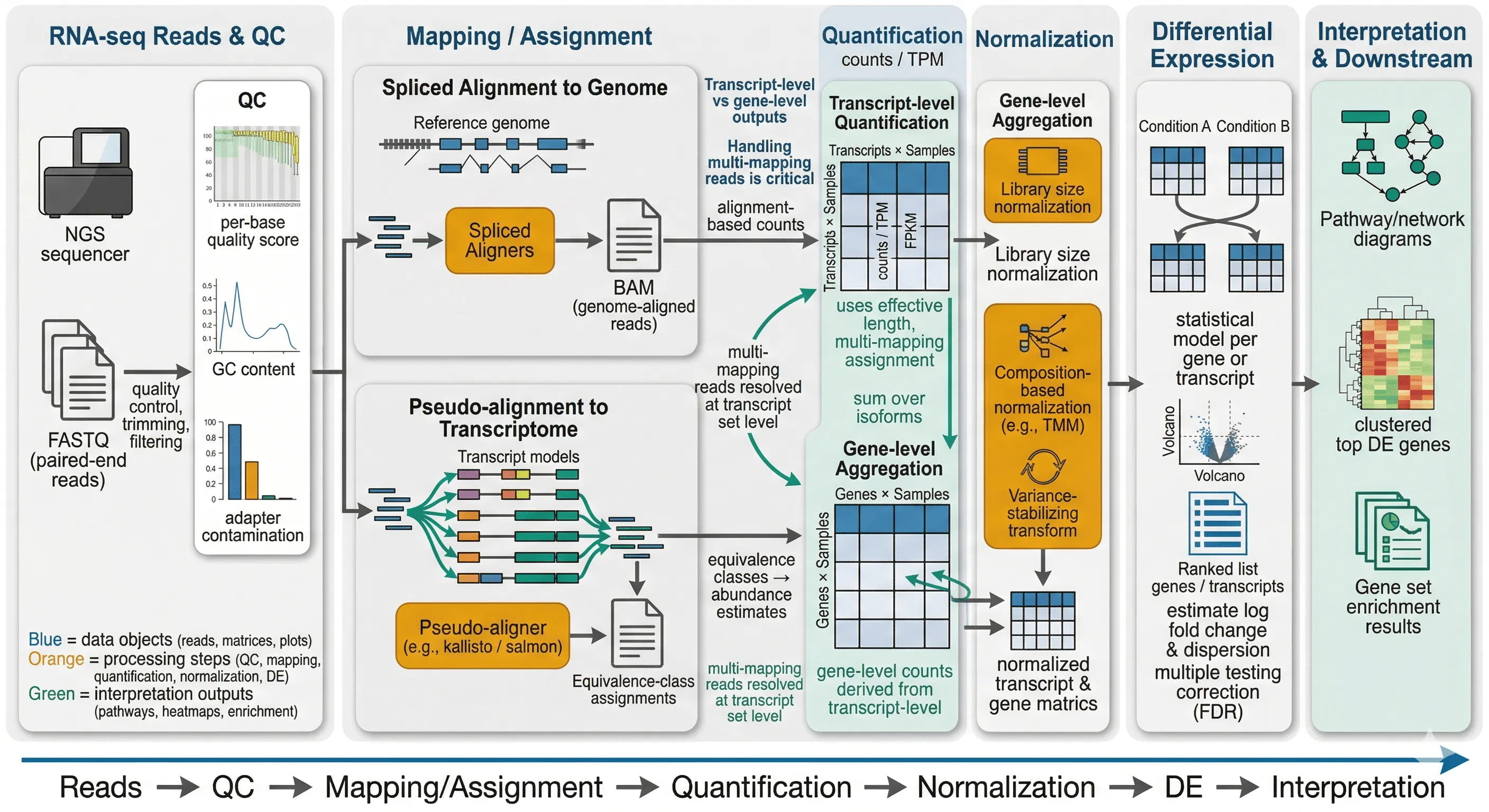
真正上手时,流程中的关键选择常常不是”用哪个工具更流行”,而是:
- 当前问题是否需要精确定位还是快速定量;
- 结果是 gene-level 还是 transcript-level;
- 是否存在明显批次效应;
- 参考和注释版本是否完全匹配。